
Pakistan’s Natural Experiment in Mendelian Genetics
How autozygosity makes recessive gene loss visible

Mendelian genetics began with an unusually clean experiment. Mendel could cross garden peas, follow traits across generations, and count the ratios. If a trait was recessive, he could make it visible by breeding plants until two copies of the relevant variant met in the same organism.
Human genetics is much messier. We cannot run breeding experiments in people, and many loss-of-function variants are so rare that they usually appear in only one copy. For autosomal genes, a person normally inherits two copies, one from the mother and one from the father. If one copy is inactive, the other may often be enough. The more revealing question is the recessive one: what happens when both copies are lost?
That is why Pakistan is such an unusually informative case. The country has a high rate of consanguineous marriage, especially cousin marriage, which increases autozygosity: the chance that both copies of a genomic segment are inherited from a shared ancestor. This does not recreate Mendel’s pea experiments by design, but it produces something closer than most human datasets. Rare variants that would usually remain hidden in heterozygous form become much more likely to appear in homozygous form.
In a paper just published in Nature, Christopher Koch, Danish Saleheen, and colleagues use this natural structure to build the Pakistan Genome Resource: exome and genome sequencing from 173,303 people recruited across Pakistan.
The authors found naturally occurring homozygous loss-of-function variants in 6,476 genes, roughly one-third of all protein-coding genes. Some of these losses are harmful. Some seem surprisingly well tolerated. Some may even be protective. For medicine, that distinction matters. A person who is naturally missing both working copies of a gene can reveal something that cell culture, mouse models, and association studies often cannot: what long-term loss of that gene looks like in a human body.
Why Pakistan?
Pakistan is useful here for two reasons.
The first is consanguinity. The country has a high rate of consanguineous marriage, especially cousin marriage, and this is exactly the demographic setting in which rare recessive variants become easier to observe. In the Pakistan Genome Resource, 30.6% of participants self-reported first-cousin parents, and the measured runs of homozygosity were consistent with elevated familial relatedness. That matters because it raises autozygosity: the chance that the two copies of a genomic segment are inherited from the same ancestor.
This makes makes Pakistan unusually informative for a specific genetic question. In a more outbred population, many rare loss-of-function variants remain hidden in heterozygous carriers. In a more autozygous cohort, some of those variants appear in homozygous form, allowing researchers to observe the consequences of complete gene loss.
The second reason is under-sampling. South Asians remain underrepresented in global genetic databases, and the study shows how much variation was missing. The authors identified 6.6 million coding variants. About 47% were absent from non-South Asian gnomAD samples, and about 30% were absent even from all of gnomAD, including South Asians.
Human knockouts
The older phrase for these individuals is “human knockouts,” but the term can mislead if taken too literally. A laboratory knockout is engineered. A person with a homozygous loss-of-function variant is not engineered; they are a naturally occurring carrier of two inactive copies of a gene. Still, the analogy is useful because drug developers are often trying to do, partially and reversibly, what genetics sometimes does completely and lifelong.
PCSK9 is the classic example. Human genetics showed that reduced PCSK9 function lowers LDL cholesterol and protects against cardiovascular disease, helping make PCSK9 inhibition an attractive therapeutic strategy. The broader logic is now central to drug development: if people who naturally lack a gene are healthy, or healthier with respect to a disease endpoint, then inhibiting that gene becomes more attractive. If they are sick, the target may be dangerous.
The Pakistan Genome Resource makes this kind of evidence much more abundant. The authors report homozygous predicted loss-of-function variants in 6,476 genes, and more than 34,000 participants carried a homozygous loss-of-function variant in at least one gene. In gnomAD, the rate of discovering such genotypes is much lower for most ancestry groups. The Amish sample is a useful comparison because it is also enriched for relatedness, but it is tiny compared with PGR. Pakistan offers both scale and autozygosity.
The result is not just a long list of rare variants. It is a way to sort genes into rough functional categories: genes where complete loss is compatible with adult life, genes where complete loss causes an obvious phenotype, genes where complete loss may be protective, and genes where homozygous loss is depleted, suggesting strong selection or essential function.
That last category is easy to overlook, but it may be one of the most important. If a large autozygous cohort rarely or never produces homozygous loss-of-function carriers for a gene, while comparable neutral variants do appear, then complete loss of that gene may be incompatible with development, survival, fertility, or recruitment into an adult cohort.
The genome’s no-fly zones
The paper uses homozygous loss-of-function variants to ask which biological systems tolerate complete disruption. The answer is reassuringly coherent. Genes essential in cell screens, genes whose mouse knockouts are embryonic lethal, genes linked to ClinGen disease annotations, broadly expressed genes, and targets of approved drugs were all depleted for homozygous loss-of-function. Developmental and core cellular pathways, including TGF-beta signaling, Notch, Hedgehog, oxidative phosphorylation, DNA repair, protein secretion, and the unfolded protein response, also showed depletion.
This is population genetics behaving like a functional screen. If a gene can be knocked out without much consequence, living adults with two inactive copies may appear in a large enough autozygous cohort. But if a gene is necessary for development, survival, fertility, or recruitment into the study, those homozygotes will be missing or strongly depleted.
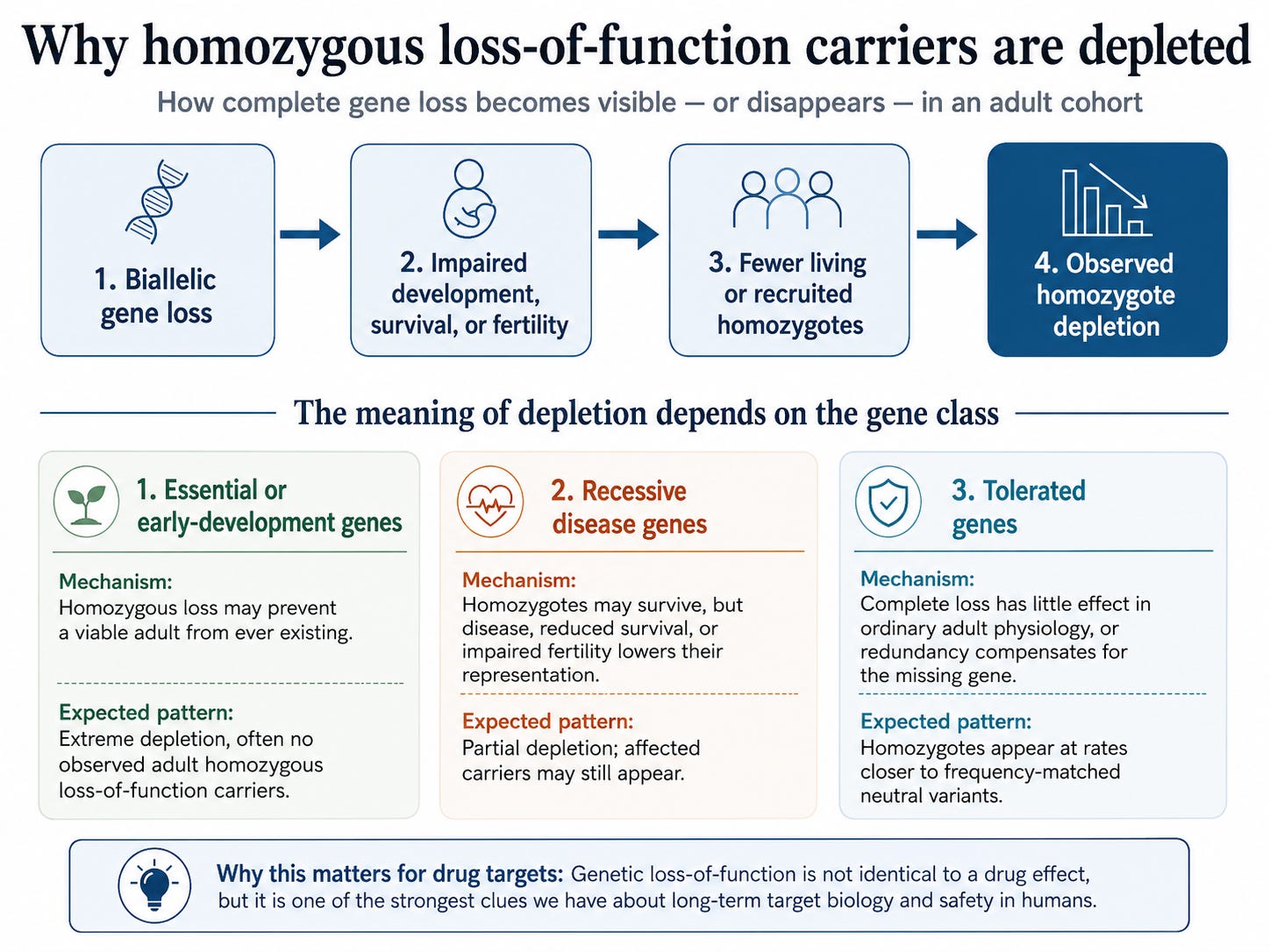
The diagram summarizes the logic, but the interpretation depends on the type of gene. For essential or early-development genes, complete loss may prevent a viable adult from ever existing. In that case, the strongest signal is not an affected adult in the dataset but the absence of homozygous loss-of-function carriers altogether. For recessive disease genes, the pattern may be less extreme. Homozygotes may survive, but disease, reduced survival, impaired fertility, or lower participation in an adult cohort can reduce their representation. For tolerated genes, complete loss has little effect in ordinary adult physiology, or biological redundancy compensates for the missing gene, so homozygotes appear at rates closer to frequency-matched neutral variants.
Drug targets are not chosen only for efficacy. They must also be safe. A genetic loss-of-function phenotype is not identical to a drug effect: a drug may be partial, reversible, tissue-specific, dose-dependent, or started late in life, while inherited gene loss is lifelong and present from conception. But human loss-of-function genetics is still one of the strongest clues we have about long-term target biology. If complete loss of a gene is well tolerated in living humans, that is useful information. If complete loss is absent from a cohort where it should otherwise be visible, that is useful information too.
The general logic is simple enough: Pakistan’s high rate of consanguineous marriage makes rare recessive gene loss visible in a way most human datasets cannot.
But the most interesting part of the paper is what appears once you look gene by gene and trait by trait. The rest of the post looks at the specific findings: which loss-of-function variants point to safer drug targets, which ones raise warning flags, why HBB shows an unexpected cholesterol signal, why LRRK2 matters for Parkinson’s drug development, and what the paper finds when it links inbreeding itself to fertility, education, body size, cardiometabolic traits, tuberculosis, and kidney disease.
There is also a population-genetic twist: Pakistan is not a single genetic block, and one group in the dataset shows a surprising trace of recent African ancestry.